Introducing Project EQUUS and ALOE
For decades, chemists have faced a frustrating asymmetry. You can spend days of compute, or weeks of bench work, finding out whether a molecule will behave the way you hoped, and only after you have already invested the time. The thermodynamic landscape of a reaction, the map that tells you which transformations are favorable and which are dead ends, usually only comes into focus after the expensive part is done.
We kept asking a simpler question: what if you knew that landscape before you ran the reaction?
Today we are sharing our first answer. Meet Project EQUUS.
What is Project EQUUS
Project EQUUS (Effective QUest into boUnded chemical Spaces) is an ultrafast virtual screening workflow for the thermochemical properties of organic molecules. It was born from research at the Garcia-Bosch Lab at Carnegie Mellon University, in collaboration with the Isayev Group, and it is built by the same small team behind Cavall Labs.
The first iteration of EQUUS focuses on redox-active organic small molecules, particularly diamines, for applications in catalysis and redox mediation. Using the AIMNet2 and AIMNet2-NSE neural network potentials, we virtually screened more than half a million small molecules for their bond dissociation free energy (BDFE), one of the single most important parameters for understanding how these molecules behave.
Where experimental data was sparse, we did not wait for it. Phenylenediamines are a good example: they are highly relevant as ligands in organometallic reactions, yet there is a significant information gap around this family compared to what we know about C-H, O-H, and S-H bonds. So we built the map ourselves, from the ground up, producing what is, to our knowledge, the largest study on redox-active molecules to date. The full work is available as a preprint on ChemRxiv.
The result is a tool that gives chemists a read on the thermodynamic landscape of their reaction before it ever happens, with near-DFT accuracy, in seconds rather than days.
A quick word on BDFE
Bond dissociation free energy is, in essence, the energy required to homolytically break a bond between two atoms. EQUUS focuses on the average BDFE of redox-active molecules that carry two N-H, O-H, or S-H bonds positioned next to each other, the kind of chemistry at the heart of proton-coupled electron transfer (PCET). Getting fast, reliable estimates of this number is exactly what lets you rank candidate molecules and rule out the unpromising ones early.
At the heart of it all: ALOE
Powering EQUUS is ALOE, the Adaptive Lightweight Optimization Engine.
The idea behind ALOE is simple to describe and hard to do well. You give it a SMILES string, and it handles everything else: generating stereoisomers, embedding and optimizing 3D conformers, ranking them, and computing the electronic and thermochemical properties you actually care about, including Gibbs free energies. Under the hood it is powered by foundational neural network potentials like AIMNet2, with a backend adapted from Auto3D.
The full pipeline runs as a sequence of clean, composable steps: generate stereoisomers, embed conformers, optimize conformers, rank conformers, and calculate thermochemistry.
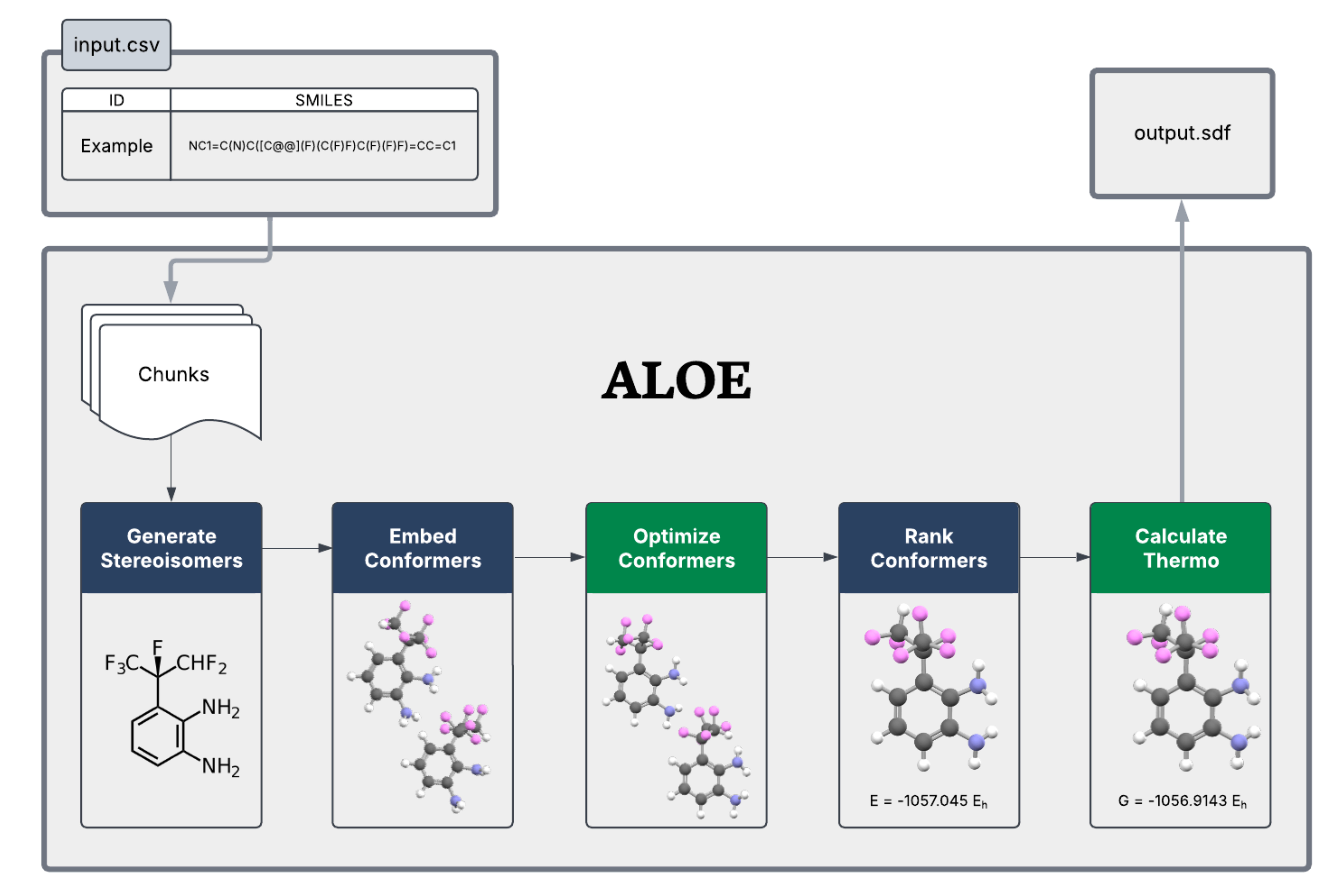
Where ALOE really shines
Two things set ALOE apart.
The first is modularity. The pipeline gives you full control over each individual operation, so you can run the whole end-to-end workflow or just the piece you need. One molecule, a thousand, or a million, it scales without changing how you work.
The second is that it runs anywhere. From a laptop to an HPC cluster, ALOE detects your available hardware and plans an optimal batched calculation strategy around it. Molecules are batched at the start of a job according to their size and your system's memory, and every step after that runs concurrently to make the most of the CPUs and GPUs you have. You get results as fast as your machine can deliver them, whatever machine that is.
It is open source, and it is yours
We built ALOE with everything we had, and today we are sharing with you. We are open-sourcing the engine under an MIT license, and the EQUUS dataset and predictions are live for anyone to explore. Getting started is one line:
pip install aloe-engineExplore the dataset, run your own predictions, break things, file issues, and send pull requests. The best tools for the next breakthrough should be in the hands of the chemists chasing it.
This is only the beginning
We are reimagining how chemistry should be done: fast, open, accurate, and intelligent. EQUUS and ALOE are our first step toward that, and there is much more to come.
Links
- Project EQUUS: cavall.chem.cmu.edu/equus
- ALOE on GitHub: github.com/EQUUS-ORG/ALOE
- Read the preprint (ChemRxiv): chemrxiv.org